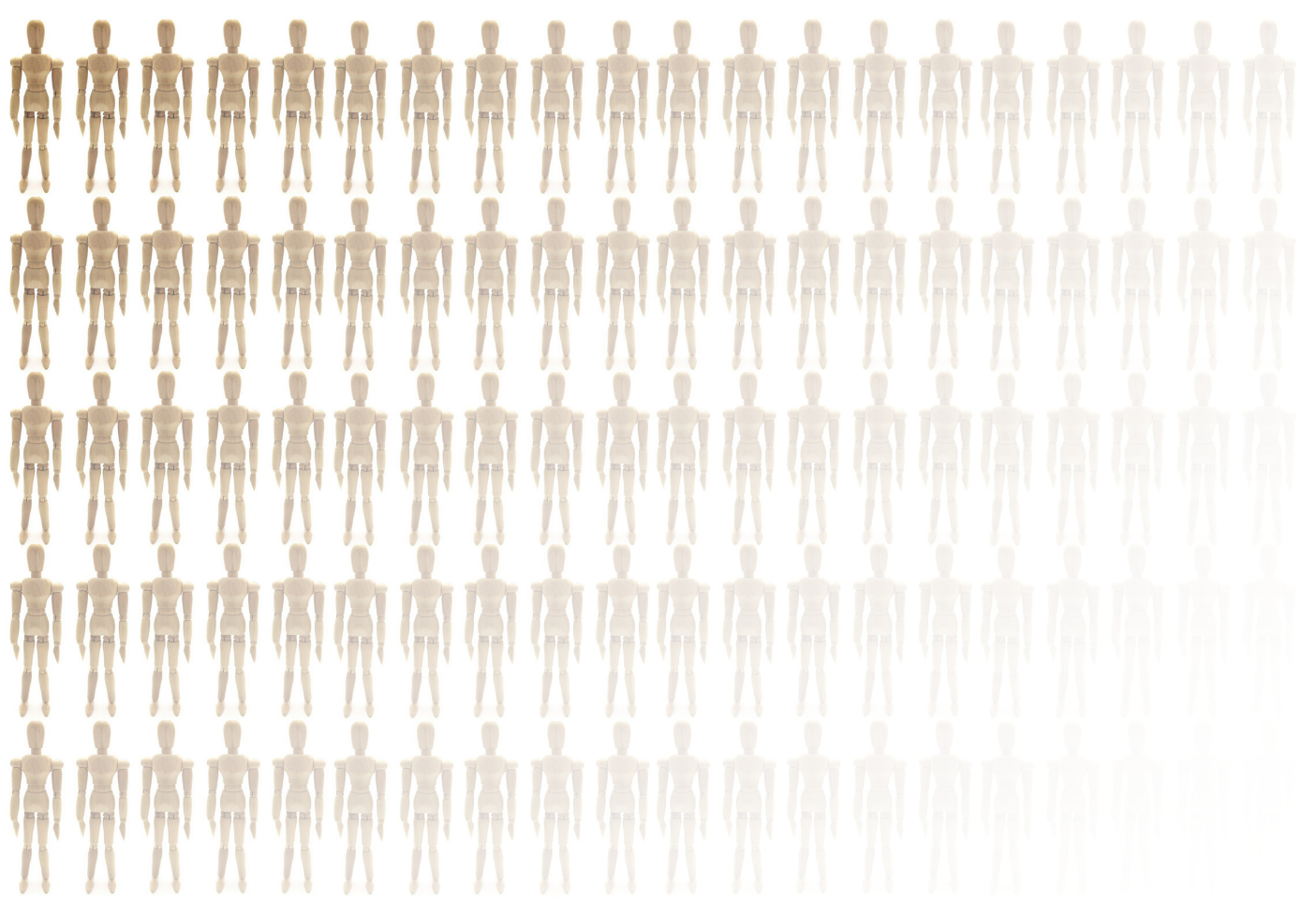
How much does developing a novel therapeutic cost? – Factors Affecting Drug Development Costs across the Pharma Industry: A mini-Report
Introduction Data evaluating the costs associated with developing novel therapeutics within the pharmaceutical industry can be used to identify trends over time and can inform more accurate budgeting for future research projects. However, the cost to develop a drug therapeutic is difficult to accurately evaluate, resulting in varying estimates ranging from hundreds of millions to billions of US dollars between…