Bayesian approach for sample size estimation and re-adjustment in clinical trials
Accurate sample size calculation plays an important role in clinical research. Sample size in this context simply refers to the number of human patients, wheather healthy or diseased, taking part in the study. Clinical studies conducted using an insufficient sample size can lack the statistical power to adequately evaluate the treatment of interest, whereas a superfluous sample size can unnecessarily waste limited resources.
Various methods can be applied for determining the optimal sample size for a specific clinical study. Methods also exist for any re-adjustments throughout the study, if required. These methods vary widely from straightforward tests and formulas to complex, time-consuming ones, depending on the type of study and available information from which to make the estimate. Most commonly used sample size calculation procedures are developed from a frequentist perspective
Importance of knowing your study parameters
Accurate sample size calculation requires, information on several key study and research parameters. These parameters usually include an effect size and variability estimate, derived from available sources; a clinically meaningful difference. In practice these parameters are generally unknown and must be estimated from the existing literature or from pilot studies.
The Bayesian Framework in sample size estimations and re-adjustments
The Bayesian Framework has gradually become one of the most frequently mentioned methods when it comes to randomised clinical trial sample size estimations and re-adjustments.
In practice, sample size calculation is usually treated explicitly as a decision problem and employs a loss or utility function.
The Bayesian approach involves three key stages:
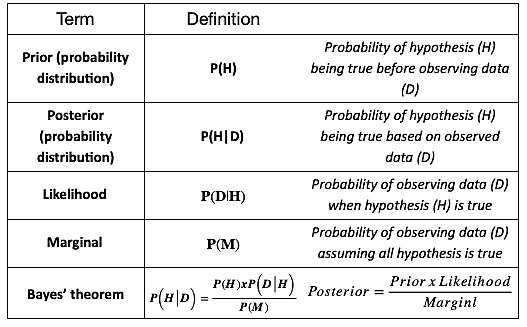
- 1. Prior estimate
A researcher has a prior estimate about the treatment effect (and other study parameters) that has been derived from meta-analysis of existing research, pilot studies, or based on expert opinion in absence of these.
- 2. Likelihood
Data is simulated to derive a likelihood estimate of prior parameters.
- 3. Posterior estimate
Based on the insights obtained, prior estimates from the first stage are updated to give a more precise final estimate.
A challenge of using this approach is knowing when to stop this cycle when enough evidence has been gathered and avoid creating bias (Dreibe,2021). Peaking at the data in order to make a stopping decision is called “optional stopping”. In general an optional stopping rule is cautioned against as it can increase type one error rates (de Heide & Grunewald, 2021).
How to decide when to stop the simulation cycle?
There are two approaches one could take.
- 1. Posterior probability
Calculating the posterior probability that the mean difference between the treatment and control arm is equal or greater than the estimated effect of the intervention. Based on the level of probably calculated (low or high) the cycle could be stopped and without any further need to gather more data.
- 2. PPOS ( predictive probability of success)
Calculating the predictive probability of achieving a successful result at the end of the study is a commonly used approach. It is really helpful when it comes to determining the success or failure of a study. Similarly, as with posterior probability based on the level of probability a decision could be made to stop or continue the study.
How to plan a Bayesian sample size calculation for a clinical trial
The key elements to consider when planning a Bayesian clinical trial are the same as for frequentists clinical trial.
Key planning stages:
- Determine the objective of the clinical study
- Determine and set endpoints
- Decide on the appropriate study design
- Run a meta analysis or review of existing evidence related to your research objective
- Statistical test and statistical analysis plan (SAP)
Even though the key planning stages are the same for both approaches it does not mean that they can be mixed through out the study. If you have chosen to use one approach you can’t change to another method once the calculations have been generated and research started.
Bayesian approach vs Frequentist approach for sample size calculations
| Bayesian | Frequentist |
| Prior and posterior( uses probability of hypothesis and data) | No prior or posterior( never gives probability of hypothesis) |
| Sample size depends on the prior and likelihood | Sample size depend on the likelihood |
| Requeres finding/deciding on prior in order to estimate sample size | Does not require prior to estimate sample size |
| Computationally intensive due to integration over many parameters | Less computationally intense |
Frequentist measures such as p-values and confidence intervals continue to predominate the methodology across life sciences research, however, the use of the Bayesian approach in sample size estimations and re-estimation for RTCs has been increasing over time.
Bayesian approach for sample size calculations in medical device clinical trial
In the recent years Bayesian approach has gained more popularity as the method used in clinical trials including medical device studies. One of the reasons being that if good prior information about the use of the specific therapeutic or device is available, the Bayesian approach may allow to include this information into the statistical analysis part of the clinical trial. Sometimes, the available prior information for a device of interest may be used as a justification for smaller sample size and shorten the length of the pivotal trial (Chen et al., 2011).
Computational algorithms and growing popularity of Bayesian approach
Bayesian statistical analysis can be computationally intense. Despite that there have been multiple breakthroughs with computational algorithms and increased computing speed that have made it much easier to calculate and build more realistic Bayesian models, further contributing to the popularity of Bayesian approach. (FDA, 2010).
Markov Chain Monte Carlo (MCMC) method
One of the basic computational tools being used is Markov Chain Monte Carlo ( MCMC) method. This method computes large number of simulations from the distributions of random quantities.
Why MCMC?
MCMC helps to deal with computational difficulties one often can face when using Bayesian approach for needed sample size estimations. The MCMC is an advanced random variable generation technique which allows one to simulate different samples from more sophisticated probability distributions.
Conclusion
Sample size calculation plays an important role in clinical research. If underestimated, statistical power for the detection of a clinically meaningful difference will likely be insufficient; if overestimated, resources are wasted unnecessarilly.
The Bayesian Framework has become quite popular approach for sample size estimation. There are advantages of using the Bayesian method, depite this there has been some criticism of this approach as a sample size estimation and re-adjustment method due to the prior being subjective and possibility of different researchers selecting different priors leading to different posteriors and final conclusions.
In reality, both the Bayesian and frequentist approaches to sample size calculation involve deriving the relevant input parameters from the literature or clinical expertise and could potentially differ due to variations in individual expert opinion as to which studies to include or exclude in this process.
Bayesian approach is more computationally intensive compared to the traditional frequentist approaches. Therefore, when it comes to selecting a method for sample size estimation, it should be chosen carefully to best fit the particular study design and base-on advice provided by statistical professionals with expertise in clinical trials.
References:
Bokai WANG, C., 2017. Comparisons of Superiority, Non-inferiority, and Equivalence Trials. [online] PubMed Central (PMC). Available at: <https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5925592/> [Accessed 28 February 2022].
Chen, M., Ibrahim, J., Lam, P., Yu, A. and Zhang, Y., 2011. Bayesian Design of Noninferiority Trials for Medical Devices Using Historical Data. Biometrics, 67(3), pp.1163-1170.
E, L., 2008. Superiority, equivalence, and non-inferiority trials. [online] PubMed. Available at: <https://pubmed.ncbi.nlm.nih.gov/18537788/> [Accessed 28 February 2022].
Gubbiotti, S., 2008. Bayesian Methods for Sample Size Determination and their use in Clinical Trials. [online] Core.ac.uk. Available at: <https://core.ac.uk/download/pdf/74322247.pdf> [Accessed 28 February 2022].
U.S. Food and Drug Administration. 2010. Guidance for the Use of Bayesian Statistics in Medical Device Clinical. [online] Available at: <https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-use-bayesian-statistics-medical-device-clinical-trials> [Accessed 28 February 2022].
van Ravenzwaaij, D., Monden, R., Tendeiro, J. and Ioannidis, J., 2019. Bayes factors for superiority, non-inferiority, and equivalence designs. BMC Medical Research Methodology, 19(1).
de Heide. R, Grunewald, P.D, 2021, Why optional stopping can be a problem for Bayesians; Psychonomic Bulletin & Review, 21(2), 201-208.